

David J. Weber, M.D., M.P.H., Division of Infectious Diseases, University of North Carolina, School of Medicine, Chapel Hill, North Carolina; Department of Hospital Epidemiology, University of North Carolina Hospitals, Chapel Hill, North Carolina; Lionel Pineau, Ph.D., president and scientific director, Eurofins Biotech Germande; corresponding author James R. Bolton, president, Bolton Photosciences Inc.
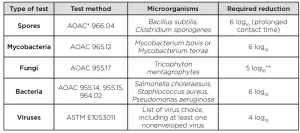
The high level of disinfection (HLD) of medical devices plays a crucial part for patient safety in healthcare. According to the US Centers for Disease Control and Prevention (CDC) guideline,1 devices that contact mucous membranes or nonintact skin (also called semi-critical devices) must be high-level disinfected. HLD is traditionally based on chemicals (liquid or gas) and normative tests used to demonstrate that their activities have been designed accordingly. According to the US Food and Drug Administration (FDA) guidance,2 HLD claim must be based on achieving certain log reduction criteria of a set of microorganisms (Table 1).
Several companies have developed ultraviolet- (UV) based systems to perform disinfection on medical devices without channels (e.g., ultrasound probes), and UV HLD brings the following substantial advantages that deserve to be proposed to the healthcare community:
- Fast reprocessing time (less than three minutes compared to chemical HLD systems that usually take up to 14 minutes).3
- Chemical-free and automatic traceability, thanks to UV sensors. Healthcare professionals have voiced their wish to use an easier approach to perform HLD and, for that matter, mention UV HLD reprocessing.4-6
Establishing a standard for UV-based HLD is highly desirable and falls well in line with the current work by the International Ultraviolet Association (IUVA) to establish standards for UV use within healthcare settings. However, the standard for UV-based HLD should be as stringent as the standard for chemical-based HLD.
Therefore, the UV HLD standard should be based on the US FDA requirements for HLD products and will differ from requirements for room disinfection. Indeed, the intention is not the same. For a room, the principal surfaces should be disinfected enough to significantly reduce environmental pathogens, such that they do not cause healthcare-associated infections (HAIs). In the case of HLD, the disinfection also is intended to eliminate all contamination that is potentially contained in body fluids to avoid patient-to-patient cross-infections.
The US FDA currently evaluates HLD chemical products with a three-tier program: potency tests, simulated use tests and in-use tests, a methodology that should be similarly followed, adapted for UV HLD and developed in a dedicated standard.
Potency tests
The FDA evaluation of an HLD product potency relies on AOAC standards using carriers of various materials and shapes. However, peni cylinders – for instance – cannot be used for UV HLD because UV isn’t meant for channeled devices or devices with interior surfaces inaccessible to light.
Here it is proposed that the UV HLD standard be based on flat carriers made of material relevant to the devices intended for reprocessing: stainless steel, ABS (acrylonitrile butadiene styrene), silicone, etc. The surface area of the inoculation should be 5 cm2, which is similar to the surface area of the peni cylinder (in and out). The inoculation should be performed on the side of the carrier that will be exposed to UV light. Several methods can be considered for inoculation: a drop that is spread or a series of droplets deposited manually, or automatically, with a pipette or a micro-pipette.
Microorganisms to consider for the tests should be those recommended by the FDA. Namely:
- spores (Bacillus subtilis, Clostridium sporogenes)
- bacteria (Staphylococcus aureus, Pseudomonas aeruginosa and Salmonella enterica)
- fungi (Trycophyton mentagrophytes)
- mycobacteria (Mycobacteria terrae)
Virucidal evaluation should be based on ASTM standards applicable with UV HLD. Viruses to be considered should be orthopoxvirus, adenovirus (Type 5), enterovirus polio and human papilloma virus, which has been recently identified as a substantial threat on which only few disinfectants are efficient.7 Test carriers must be hung at a consistent reference position in the disinfection chamber throughout the tests.
Simulated use test
A simulated use test should be performed to assess the disinfection capability of the UV HLD system on real devices. “Simulated use tests help determine factors that prevent or limit contacts and effectiveness of the germicide. Simulated use tests are controlled laboratory tests that allow the precise application of a specified and quantified inoculum to selected device surfaces.”8 Interpreting this FDA guidance:
Different device types must be tested, representative of the whole device family, from various manufacturers. For instance, in case of ultrasound probe types, endocavitary and external probes must be considered. In addition, different geometry varieties, such as hole, flat surface, groove, etc., must be included in the test. This geometrical challenge is of the upmost importance for UV HLD systems. It is proposed to name “cold spot” the area of the instrument that receives the least irradiancea in the UV chamber during a cycle. The location and relative challenge of this cold spot depends on the interaction of the medical device geometry, its position within the chamber and the UV HLD chamber configuration (number and nature of UV sources, dimensions, etc.). The inoculated areas should include the cold spot of the instrument.
For the inoculum, Mycobacterium terrae, as the most resistant mycobacterium strain, is an indicated choice for UV and should be associated with organic and inorganic challenges (e.g. 5% BSA and hard water). The expected performance is a full 6 log10 inactivation of mycobacteria.
In-use test
The role of the in-use test is to verify that the HLD performs as expected in the clinical setting. It should be recommended to have a sufficient number of samples to demonstrate the efficacy of the process in real life conditions.
Additional requirement for a UV HLD system
UV HLD systems manufacturers must have a methodology to identify and value the cold spot of the instruments intended to be reprocessed. A UV HLD system must ensure the delivery of a sufficient UV dose to reach HLD on the cold spot of the instrument during the disinfection cycle. This methodology should be verified by physical optical measurements and microbiological tests.
Conclusion
This article has laid out the requirements that a UV HLD system should test against to demonstrate it has an HLD status according to US FDA. Given the substantial advantages of UV HLD for the healthcare community, it is suggested that the concerned parties collaborate to establish a standard protocol allowing international diffusion of this technology.
Contact: jbolton@boltonuv.com; dweber@unch.unc.edu
References
- Guideline for Disinfection and Sterilization in Healthcare Facilities, 2008, CDC.
- Content and Format of Premarket Notification [510(k)] Submissions for Liquid Chemical Sterilants/High Level Disinfectants – Guidance for Industry and FDA Reviewers, 2000, FDA.
- Johnson, J., Proctor, M., Bluth, E., Smetherman, D., Baumgarten, K., Troxclair, L., and Bienvenu, M. 2013, Evaluation of a Hydrogen Peroxide-Based System for High-Level Disinfection of Vaginal Ultrasound Probes, J. Ultrasound Med. 32(10): 1799–1804.
- Talan, D.A. and Partida, C.N. 2011, Emergency Department Ultrasound Infection Control: Do Unto (and Into) Others; Annals Emerg. Med. 58(1), 64–66.
- Shokoohi, H., Armstrong, P., and Tansek, R. 2015, Emergency department ultrasound probe infection control: challenges and solutions, Open Access Emerg. Med. 7: 1–9.
- Häggström, M., Spira, J., and Edelstam, G. 2014, Transducer Hygiene: Comparison of Procedures for Decontamination of Ultrasound Transducers and Their Use in Clinical Practice, J. Clin. Ultrasound, 43(2):81–88.
- Meyers J., Rindock E., Conway M.J., Meyers C., Robison R. 2014, Susceptibility of high-risk human papillomavirus type 16 to clinical disinfectants, J Antimicrob Chemother 2014; 69: 1546–1550.
- Content and Format of Premarket Notification [510(k)] Submissions for Liquid Chemical Sterilants/ High Level Disinfectants, January 3, 2000. U.S. Department Of Health And Human Services Food and Drug Administration Center for Devices and Radiological Health, January, 2000.